Transcriptomic Analysis
Gepoliano Chaves
October 14th, 2024
- Summary
- Data
- References
- Load Libraries
- Load Gene Expression Dataframe
- Prepare enrichment set including several gene sets
- Session Information
Summary
In this part we do transcriptome analysis of RNA-Seq isolated from tumors of neuroblastoma patients. We conduct the analysis according to reference documentation.
Data
Data is available in this Google folder: https://drive.google.com/drive/u/1/folders/1lSzUkQdBJw6uQ4uXW0IFlQF_pPWkjMkH
References
This was the google search: deseq2 tutorial in r and the following references showed up:
This was the main reference I used: https://introtogenomics.readthedocs.io/en/latest/2021.11.11.DeseqTutorial.html for the pipeline itself.
This reference has a good explanation of the method DESeq2 uses for read (count) normalization. Normalization by division by the size factors or normalization factors: https://rdrr.io/bioc/DESeq2/man/varianceStabilizingTransformation.html
This is the Bioconductor reference for DESeq2: https://bioconductor.org/packages/devel/bioc/vignettes/DESeq2/inst/doc/DESeq2.html
In this other google search: RNA-Seq tutorialm these were the references:
To see how RNA-Seq is applied in the health system, I did this google search: RNA-Seq sistema unico de saude brasil and these articles were retrieved:
Pesquisa translacional na era pós-genômica: avanços na área da transcriptômica (Pacheco 2019)
Load Libraries
library(readxl)
library(DESeq2)
library(ggplot2)
library(dplyr)
library(expss)
library("clusterProfiler")
library(pheatmap)
library(RColorBrewer)
Load Gene Expression Dataframe
Reference Values: 70 samples and 300 genes
r2_gse62564_GSVA_Metadata <- readRDS( "../../SIP/SIP2023_BME03/recombio bookdown/results/r2_gse62564_GSVA_Metadata.rds")
In this part we can select the number of tumor samples to analyze. In total, the original dataframe has 498 tumor samples so for the sake of time, we will select around 50 samples to start with.
set.seed(12345)
r2_gse62564_GSVA_Metadata <- r2_gse62564_GSVA_Metadata[sample(nrow(r2_gse62564_GSVA_Metadata), size=55), ]
It is not the first time that I encounter this Error: unable to find an inherited method for function ‘select’ for signature ‘x = “data.frame”’. Then I have to write dplyr:: in front of the select function, to select the variables that will define the metadata dataframe:
r2_gse62564_Metadata <- r2_gse62564_GSVA_Metadata %>% dplyr::select(c(high_risk, mycn_status))
r2_gse62564_Metadata$high_risk <- as.factor(r2_gse62564_Metadata$high_risk)
r2_gse62564_Metadata$sampleID <- rownames(r2_gse62564_Metadata)saveRDS(r2_gse62564_Metadata, file = "./data/r2_gse62564_Metadata.rds")
In this part I transpose the metadata to allow removing the long list of phenotype scores from age_at_diagnosis to WEINMANN_ADAPTATION_TO_HYPOXIA_UP:
r2_gse62564_selected <- t(r2_gse62564_GSVA_Metadata %>% dplyr::select(-c(age_at_diagnosis:WEINMANN_ADAPTATION_TO_HYPOXIA_UP)))
Select random sample of three rows from data frame:
set.seed(12345)
r2_gse62564_rand_df <- r2_gse62564_selected[sample(nrow(r2_gse62564_selected), size=400), ]
## Make Counts Numeric and make sure they are integers
r2_gse62564_rand_num_df <- as.data.frame(r2_gse62564_rand_df)
r2_gse62564_rand_num_df <- r2_gse62564_rand_num_df %>%
mutate_if(is.character,as.numeric)%>%
mutate_if(is.numeric, round)
Read r2_gse62564_rand_num_df object
r2_gse62564_rand_num_df <- readRDS("./data/r2_gse62564_rand_num_df.rds")
Load Metadata object
r2_gse62564_Metadata <- readRDS("./data/r2_gse62564_Metadata.rds")
Construct dds object neuroblastoma
dds_nb <- DESeqDataSetFromMatrix(countData = r2_gse62564_rand_num_df,
colData = r2_gse62564_Metadata,
design = ~ high_risk)## converting counts to integer mode
Pre-filtering
keep_nb <- rowSums(counts(dds_nb)) >= 10
dds_nb <- dds_nb[keep_nb,]
Note on factor levels
dds_nb$high_risk <- factor(dds_nb$high_risk, levels = c("yes","no"))
Differential expression analysis
Genes differentially expressed between conditions
dds_nb <- DESeq(dds_nb)## estimating size factors## estimating dispersions## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimates## fitting model and testing## -- replacing outliers and refitting for 5 genes
## -- DESeq argument 'minReplicatesForReplace' = 7
## -- original counts are preserved in counts(dds)## estimating dispersions## fitting model and testingres_nb <- results(dds_nb)
res_nb## log2 fold change (MLE): high risk no vs yes
## Wald test p-value: high risk no vs yes
## DataFrame with 400 rows and 6 columns
## baseMean log2FoldChange lfcSE stat pvalue
## <numeric> <numeric> <numeric> <numeric> <numeric>
## MLK7-AS1 1.31447 -0.5075966 0.365353 -1.389331 1.64732e-01
## ZNF562 31.53441 -0.5581062 0.104586 -5.336351 9.48357e-08
## PITX1 2.74169 -1.8298833 0.467518 -3.914040 9.07644e-05
## LOC100506258 2.97734 0.1881946 0.287578 0.654413 5.12846e-01
## SRSF10 51.81808 -0.0426755 0.101486 -0.420508 6.74114e-01
## ... ... ... ... ... ...
## HIST2H4B 5.88411 -0.851124 0.246887 -3.447422 0.000565965
## RHPN1 8.96054 0.213377 0.320608 0.665539 0.505705816
## PKN2 34.02251 0.230331 0.102567 2.245675 0.024724850
## MEP1A 1.31634 0.425583 0.407642 1.044012 0.296479728
## FBXL18 44.22974 0.319733 0.128816 2.482082 0.013061717
## padj
## <numeric>
## MLK7-AS1 3.01491e-01
## ZNF562 3.06635e-06
## PITX1 6.60311e-04
## LOC100506258 6.98882e-01
## SRSF10 8.24232e-01
## ... ...
## HIST2H4B 0.0028894
## RHPN1 0.6974426
## PKN2 0.0648192
## MEP1A 0.4638473
## FBXL18 0.0383935
Log fold change shrinkage for visualization and ranking
resultsNames(res_nb)## character(0)summary(res_nb)##
## out of 400 with nonzero total read count
## adjusted p-value < 0.1
## LFC > 0 (up) : 70, 18%
## LFC < 0 (down) : 50, 12%
## outliers [1] : 0, 0%
## low counts [2] : 109, 27%
## (mean count < 1)
## [1] see 'cooksCutoff' argument of ?results
## [2] see 'independentFiltering' argument of ?results
Volcano Plot
res_nb <- res_nb[complete.cases(res_nb),]
res_nb_df <- as.data.frame(res_nb)
res_nb_volcPlot_df <- res_nb_df %>%
dplyr::mutate(differential_expression = ifelse(res_nb_df$log2FoldChange > 0 & res_nb_df$padj < 0.05, "HR Up", ifelse(res_nb_df$log2FoldChange < 0 & res_nb_df$padj < 0.05, "HR Down", "Not Significant")))
ggplot(data=res_nb_volcPlot_df,aes(x=log2FoldChange,y=-log10(padj)))+
geom_point()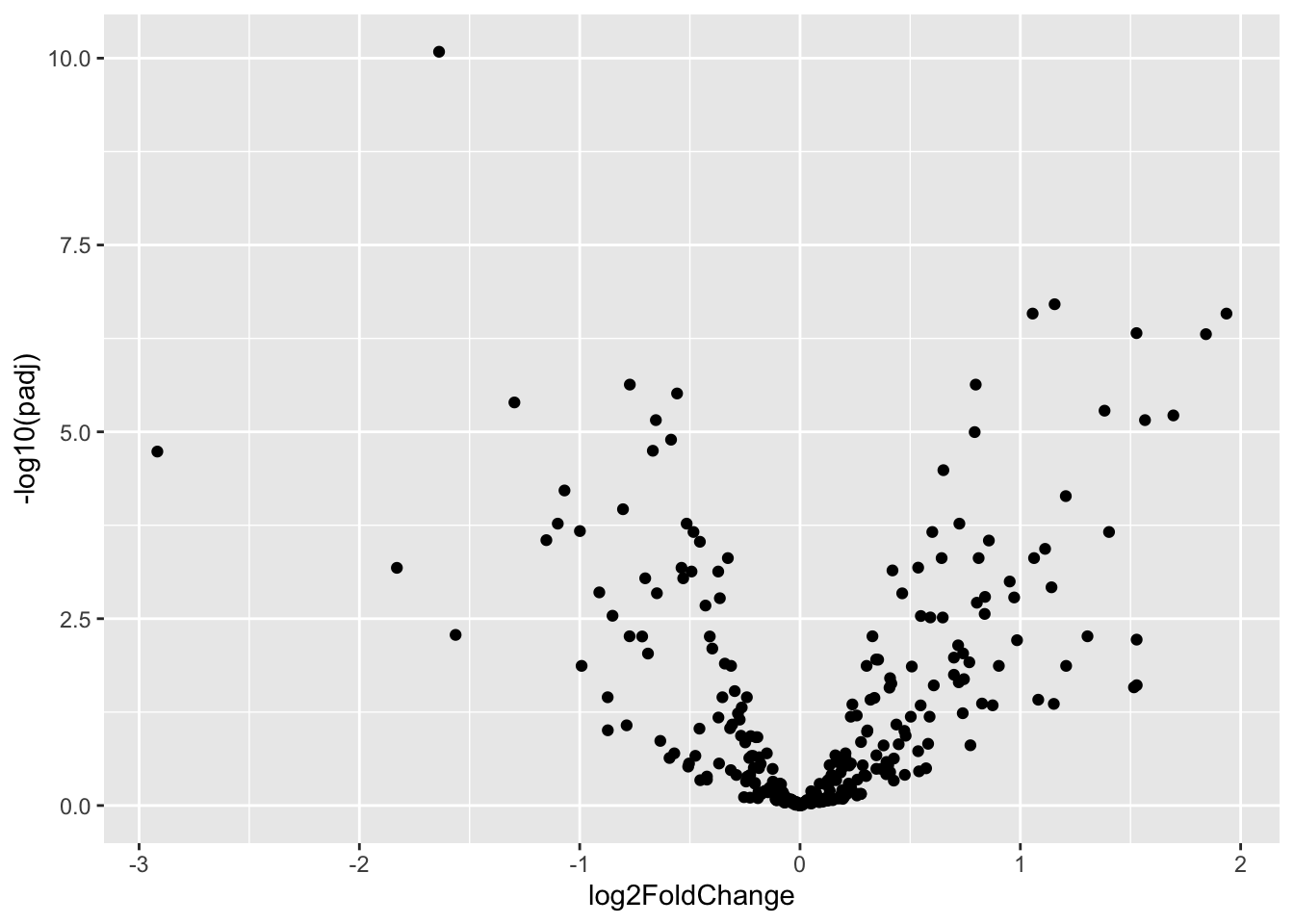
ggplot(data=res_nb_volcPlot_df,aes(x=log2FoldChange,y=-log10(padj),col=differential_expression))+
geom_point()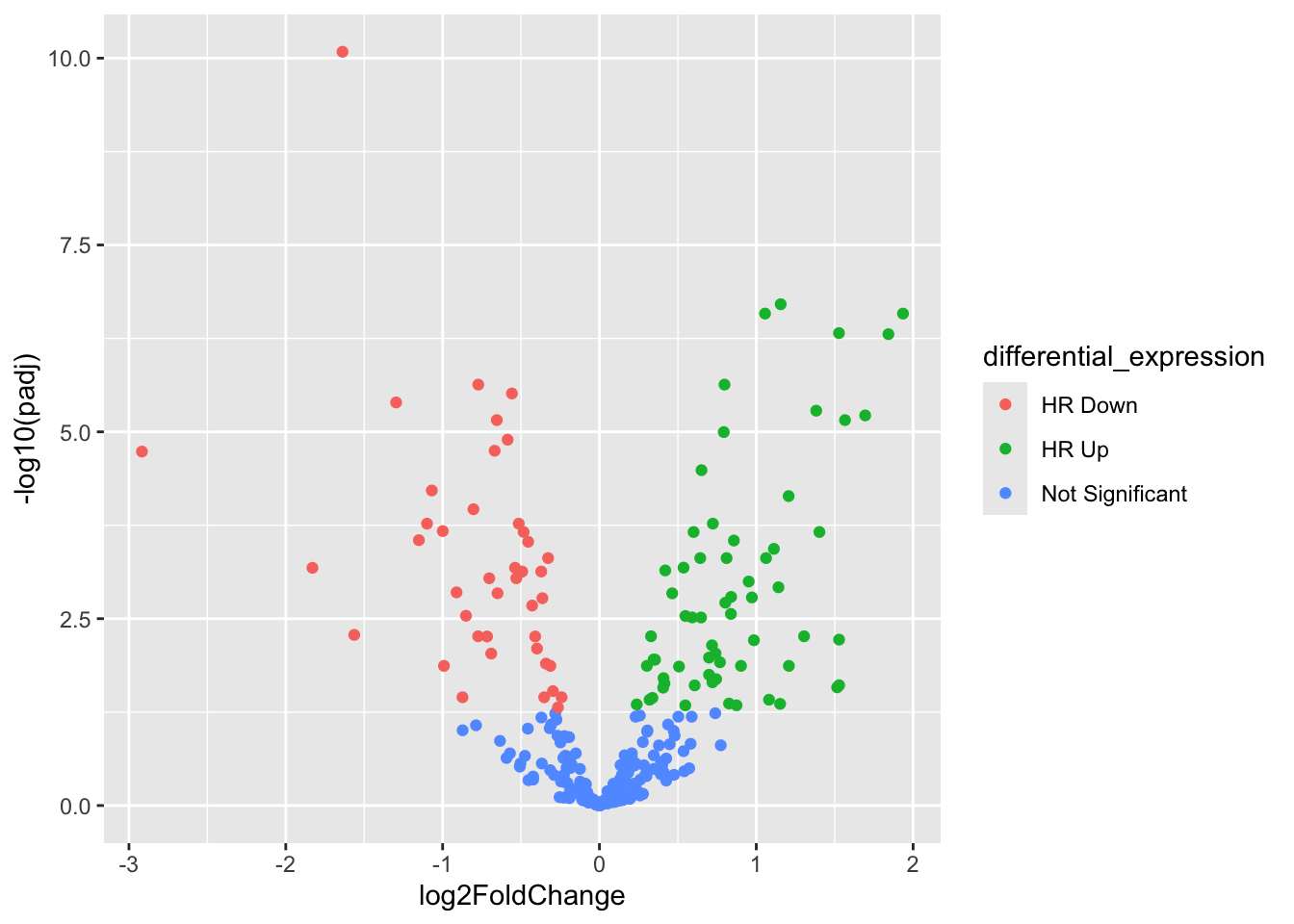
ggplot(data=res_nb_volcPlot_df,
aes(x=log2FoldChange,y=-log10(padj),col=differential_expression
))+
geom_point()+
ggtitle("Plot of HR Up/Dn") +
theme(
plot.title = element_text(color="red", size=14, face="bold.italic"),
)+
theme_bw() + # Select theme with a white background
theme(panel.border = element_rect(colour = "black", fill = NA, size= 0.5),
panel.grid.minor = element_blank(),
panel.grid.major = element_blank()) ## Warning: The `size` argument of `element_rect()` is
## deprecated as of ggplot2 3.4.0.
## ℹ Please use the `linewidth` argument instead.
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to
## see where this warning was generated.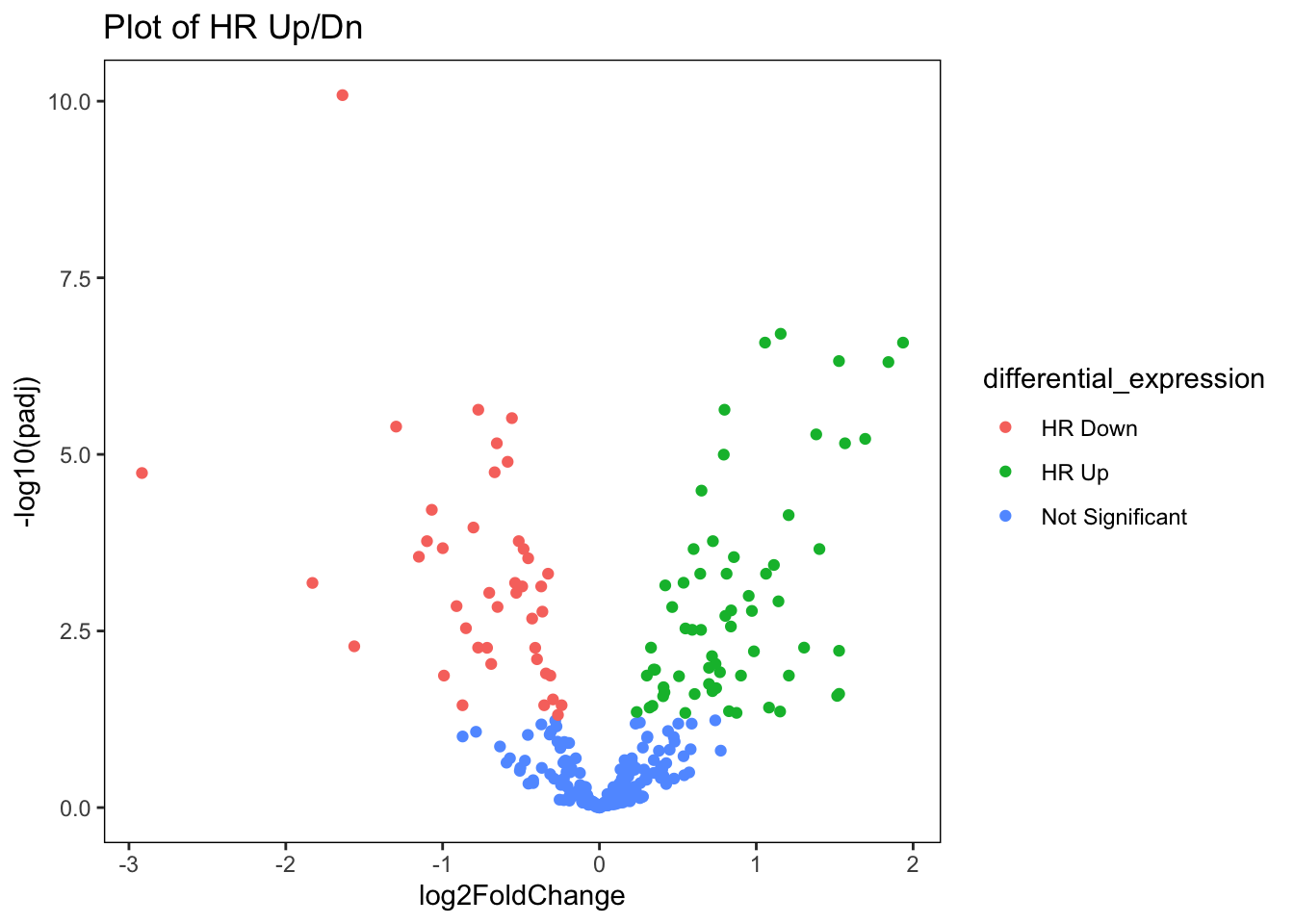
ggplot(data = res_nb_volcPlot_df,
aes(x = log2FoldChange,
y = -log10(padj))) +
geom_point(aes(colour = differential_expression),
alpha = 1,
shape = 16,
size = 1) +
geom_hline(yintercept = -log10(0.05),
linetype = "dashed") +
geom_vline(xintercept = c(-2, 2),
linetype = "dashed") +
scale_x_continuous(limits = c(-4, 4)) +
# Add y limits
scale_y_continuous(limits = c(-2, 10)) + # Have tested the y limit up to 200
# Colors of ADRN and MES Genes
scale_color_manual(values = c("#0000ff", # Blue
"#ff0000", # Gray
"#c9c9c9"))+ # Red
ggtitle("Plot of HR Up/Dn") +
theme(
plot.title = element_text(color="red", size=14, face="bold.italic"),
)+
theme_bw() + # Select theme with a white background
theme(panel.border = element_rect(colour = "black", fill = NA, size= 0.5),
panel.grid.minor = element_blank(),
panel.grid.major = element_blank()) ## Warning: Removed 1 row containing missing values or
## values outside the scale range (`geom_point()`).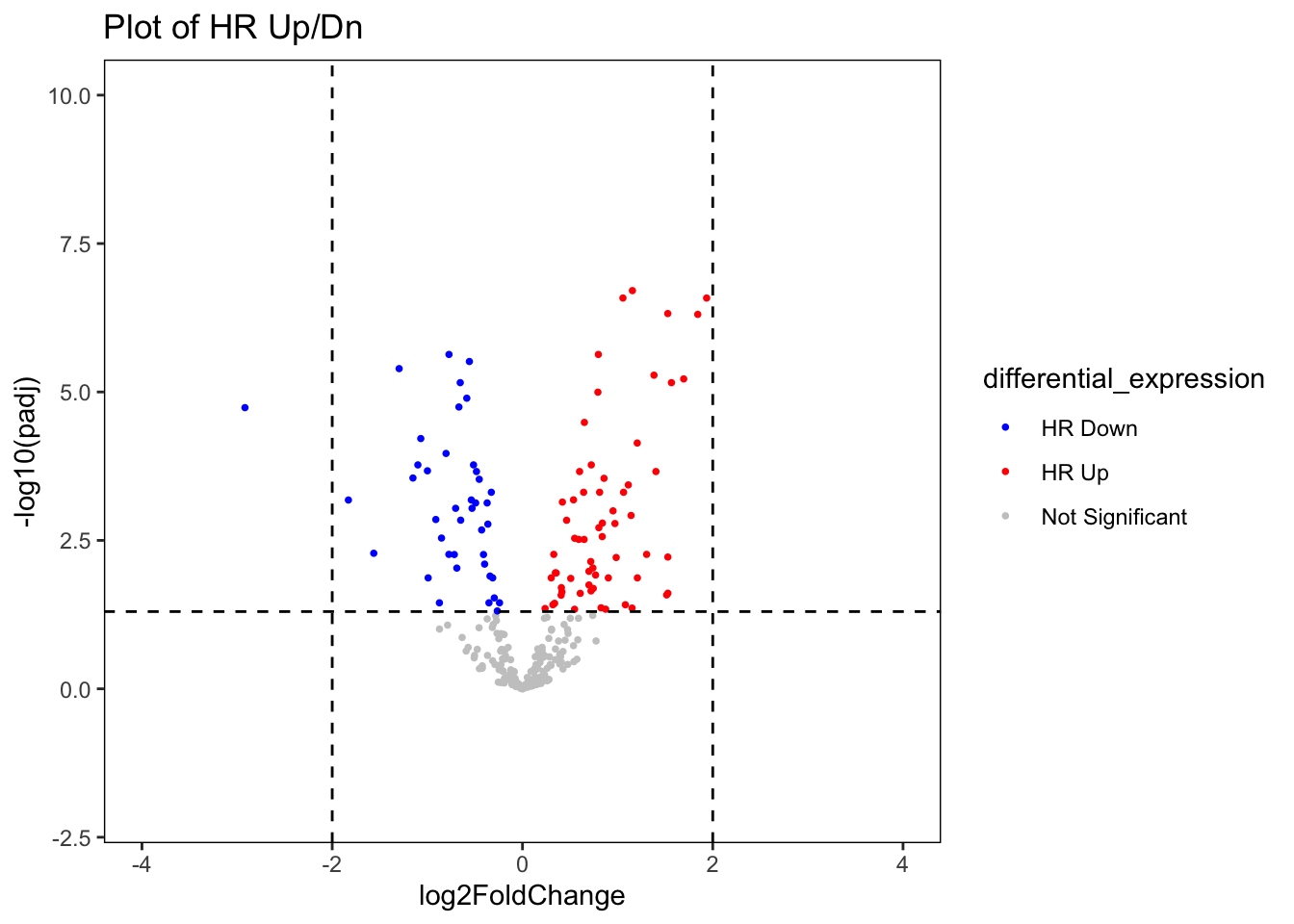
Enrichment analysis
cross_cases(res_nb_volcPlot_df, differential_expression)| #Total | |
|---|---|
| differential_expression | |
| HR Down | 42 |
| HR Up | 63 |
| Not Significant | 186 |
| #Total cases | 291 |
List of up-regulated genes
res_nb_volcPlot_df["gene_symbol"] <- rownames(res_nb_volcPlot_df)
nb_volcPlo_Up <- subset(res_nb_volcPlot_df, differential_expression == "HR Up" )
List_nb_volcPlo_Up <- as.character(nb_volcPlo_Up$gene_symbol)
length(List_nb_volcPlo_Up)## [1] 63
library(msigdbr)
h_gene_sets_H = msigdbr(species = "Homo sapiens", category = c("H"))
h_gene_sets_C2 = msigdbr(species = "Homo sapiens", category = c("C2"))
h_gene_sets_C5 = msigdbr(species = "Homo sapiens", category = c("C5"))
Prepare enrichment set including several gene sets
msigdbr_df <- rbind(h_gene_sets_H, h_gene_sets_C2)
msigdbr_df <- rbind(msigdbr_df, h_gene_sets_C5)
Define database for Enrichr to look at
msigdbr_t2g = msigdbr_df %>% dplyr::distinct(gs_name, gene_symbol) %>% as.data.frame()
ADRN Enrichment Table RNA-Seq: Up-regulated Enrichments
enrichr_nb_volcPlo_Up <- enricher(List_nb_volcPlo_Up, TERM2GENE = msigdbr_t2g)
enrichr_nb_volcPlo_Up_df <- as.data.frame(enrichr_nb_volcPlo_Up)
dotplot
dotplot(enrichr_nb_volcPlo_Up,
font.size = 6,
showCategory=10) + ggtitle("High Risk Up-Regulated")+
theme(axis.text.y = element_text(size = 6),
legend.text=element_text(size=6),
legend.key.size = unit(0.2, 'cm'))+
theme_classic()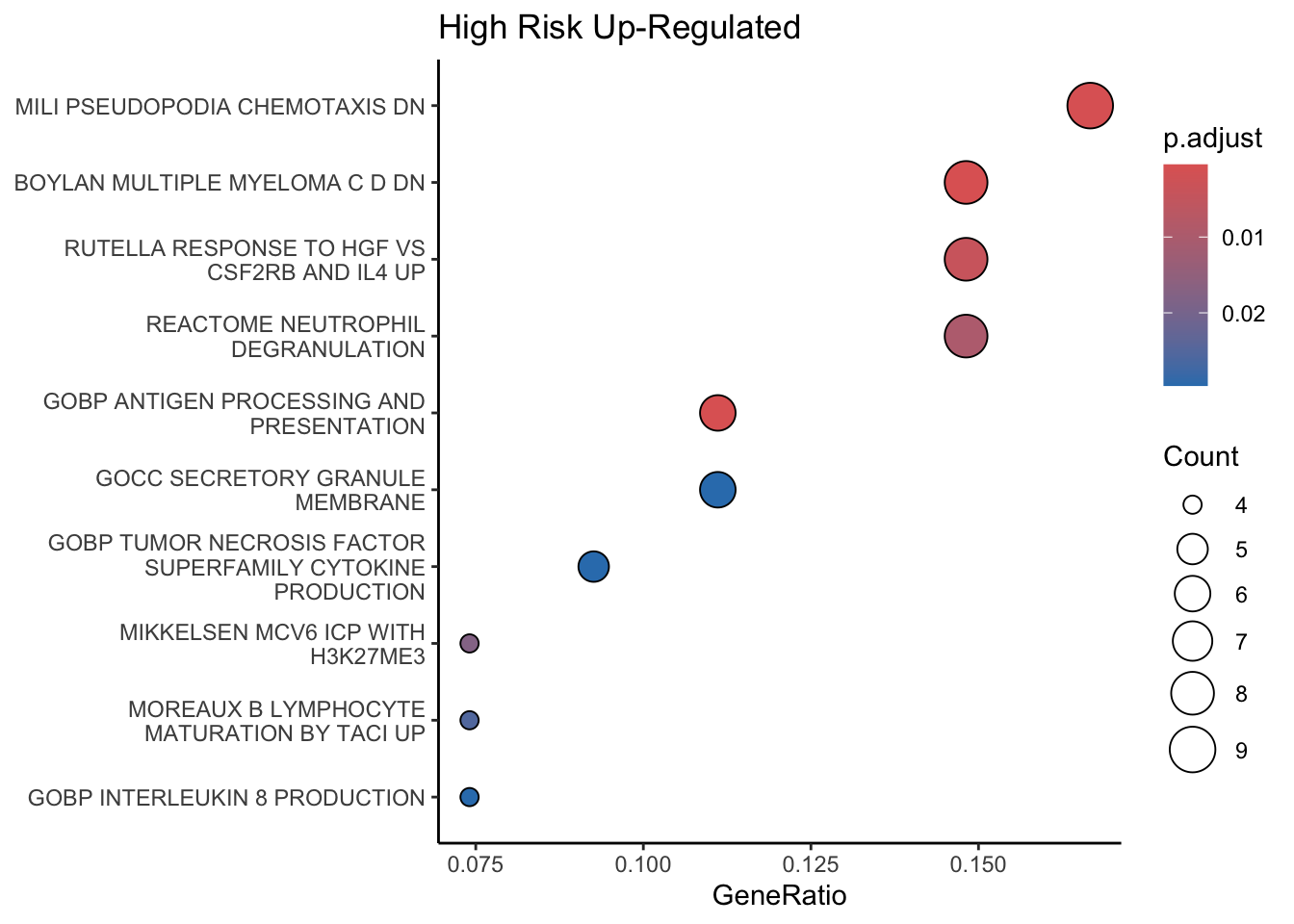
Data transformations and visualization
Extracting transformed counts values
vsd_nb <- varianceStabilizingTransformation(dds_nb)
head(assay(vsd_nb), 4)## GSM1205379 GSM1205288 GSM1205445 GSM1205455 GSM1205457 GSM1205389
## MLK7-AS1 2.688023 2.688023 2.688023 2.688023 2.977123 2.688023
## ZNF562 5.225106 5.185561 5.185561 4.872369 4.821297 4.821297
## PITX1 2.688023 2.688023 2.688023 2.688023 2.688023 2.688023
## LOC100506258 2.977123 3.192668 3.192668 2.977123 2.977123 2.977123
## GSM1205551 GSM1205330 GSM1205312 GSM1205589 GSM1205453 GSM1205495
## MLK7-AS1 2.688023 2.977123 2.688023 2.688023 2.688023 2.977123
## ZNF562 5.474709 5.507034 5.144874 4.655711 5.225106 5.373070
## PITX1 2.688023 2.688023 2.688023 2.688023 2.688023 2.688023
## LOC100506258 3.369538 2.977123 3.775873 2.688023 3.521428 2.688023
## GSM1205323 GSM1205727 GSM1205275 GSM1205340 GSM1205587 GSM1205503
## MLK7-AS1 2.688023 2.977123 2.688023 2.688023 2.737003 2.985519
## ZNF562 5.225106 5.896159 5.144874 4.325578 5.168947 5.559645
## PITX1 2.688023 2.688023 2.688023 3.775873 2.737003 4.182891
## LOC100506258 3.655450 3.521428 3.369538 3.521428 3.459248 2.985519
## GSM1205702 GSM1205653 GSM1205277 GSM1205660 GSM1205531 GSM1205523
## MLK7-AS1 2.978256 2.688023 2.688023 2.688023 2.688023 2.688023
## ZNF562 5.798292 5.225106 4.768274 5.301022 5.301022 5.144874
## PITX1 2.688848 4.325578 2.688023 2.688023 2.688023 2.688023
## LOC100506258 2.688848 2.977123 2.977123 3.369538 3.192668 2.688023
## GSM1205494 GSM1205697 GSM1205449 GSM1205565 GSM1205249 GSM1205496
## MLK7-AS1 2.977123 2.977123 2.692495 2.977123 2.690371 2.692495
## ZNF562 5.944008 5.599821 5.278415 5.686930 4.976662 5.811177
## PITX1 2.688023 3.192668 2.692495 5.373070 2.690371 2.692495
## LOC100506258 2.977123 3.192668 3.530402 2.977123 2.980348 2.692495
## GSM1205630 GSM1205251 GSM1205378 GSM1205429 GSM1205385 GSM1205343
## MLK7-AS1 2.688023 2.688023 2.688023 2.688023 2.688023 2.688023
## ZNF562 5.144874 4.768274 5.263574 5.373070 4.655711 5.225106
## PITX1 2.688023 2.688023 2.688023 2.688023 2.688023 2.977123
## LOC100506258 3.986316 2.977123 3.192668 2.977123 3.885506 3.192668
## GSM1205612 GSM1205253 GSM1205317 GSM1205299 GSM1205591 GSM1205360
## MLK7-AS1 2.688023 2.688023 2.984880 2.977123 2.688023 2.688023
## ZNF562 5.059793 5.686930 5.734612 4.872369 5.263574 5.225106
## PITX1 2.688023 2.688023 2.984880 2.688023 2.977123 2.688023
## LOC100506258 3.775873 3.192668 2.693671 2.977123 3.192668 2.977123
## GSM1205297 GSM1205598 GSM1205525 GSM1205262 GSM1205273 GSM1205469
## MLK7-AS1 2.688023 2.982874 2.688023 2.688023 2.688023 2.688023
## ZNF562 5.144874 5.644000 4.821297 4.821297 4.969218 4.821297
## PITX1 2.688023 2.692210 2.688023 2.688023 2.688023 2.688023
## LOC100506258 2.688023 2.692210 3.369538 2.977123 2.977123 3.521428
## GSM1205403 GSM1205374 GSM1205626 GSM1205448 GSM1205382 GSM1205620
## MLK7-AS1 2.977123 2.977123 2.977123 2.688023 2.977123 2.983266
## ZNF562 5.185561 5.474709 5.871620 5.185561 5.015237 5.862536
## PITX1 2.688023 2.688023 7.552517 2.688023 2.688023 5.239866
## LOC100506258 2.977123 2.688023 2.977123 2.977123 3.369538 2.983266
## GSM1205683
## MLK7-AS1 2.688023
## ZNF562 5.373070
## PITX1 2.688023
## LOC100506258 2.977123
Data quality assessment by sample clustering and visualization
select_nb <- order(rowMeans(counts(dds_nb,normalized=TRUE)),
decreasing=TRUE)[1:20]
df_nb <- as.data.frame(colData(dds_nb)[,c("high_risk", "mycn_status")])
# pheatmap(assay(dds_nb)[select_nb,], cluster_rows=FALSE, show_rownames=FALSE,
# cluster_cols=FALSE, annotation_col=df_nb)
pheatmap(assay(dds_nb)[select_nb,],annotation_col=df_nb)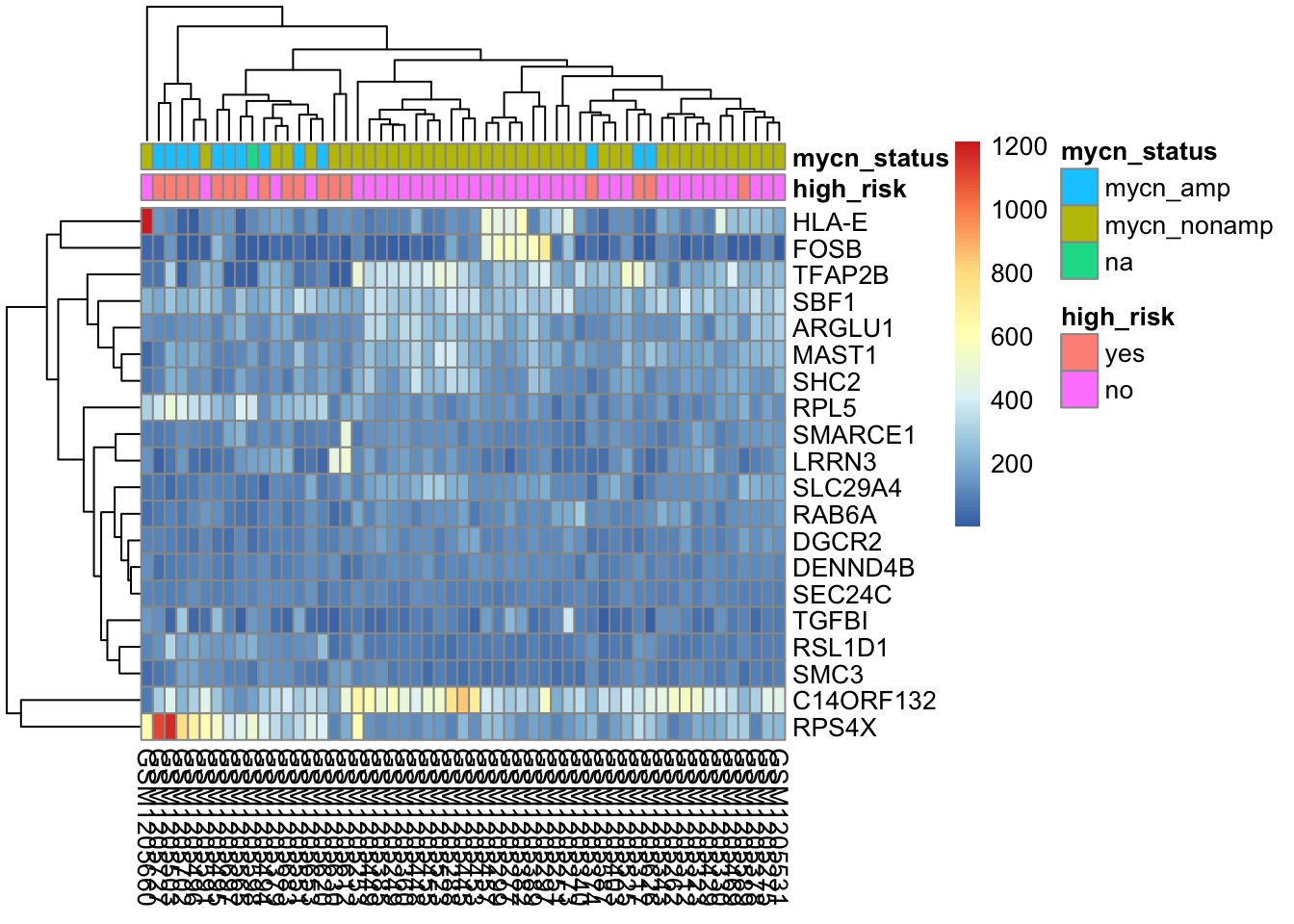
Heatmap of the sample-to-sample distances
sampleDists_nb <- dist(t(assay(vsd_nb)))sampleDistMatrixNb <- as.matrix(sampleDists_nb)
rownames(sampleDistMatrixNb) <- paste(vsd_nb$high_risk, sep="-")
colnames(sampleDistMatrixNb) <- NULL
colors <- colorRampPalette( rev(brewer.pal(9, "Blues")) )(255)
pheatmap(sampleDistMatrixNb,
clustering_distance_rows=sampleDists_nb,
clustering_distance_cols=sampleDists_nb,
col=colors)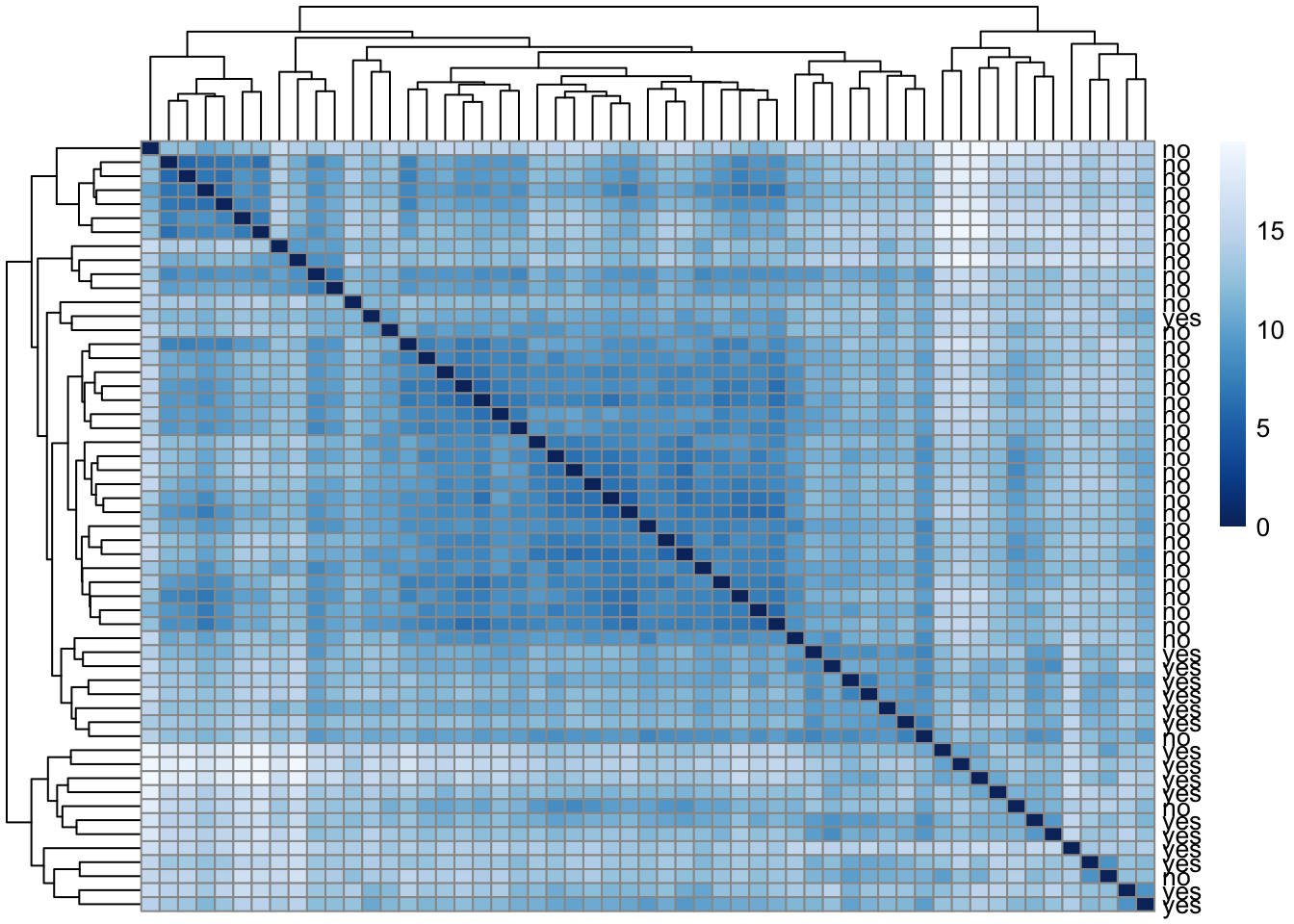
Principal component plot of the samples
plotPCA(vsd_nb, intgroup=c("high_risk", "mycn_status"))## using ntop=500 top features by variance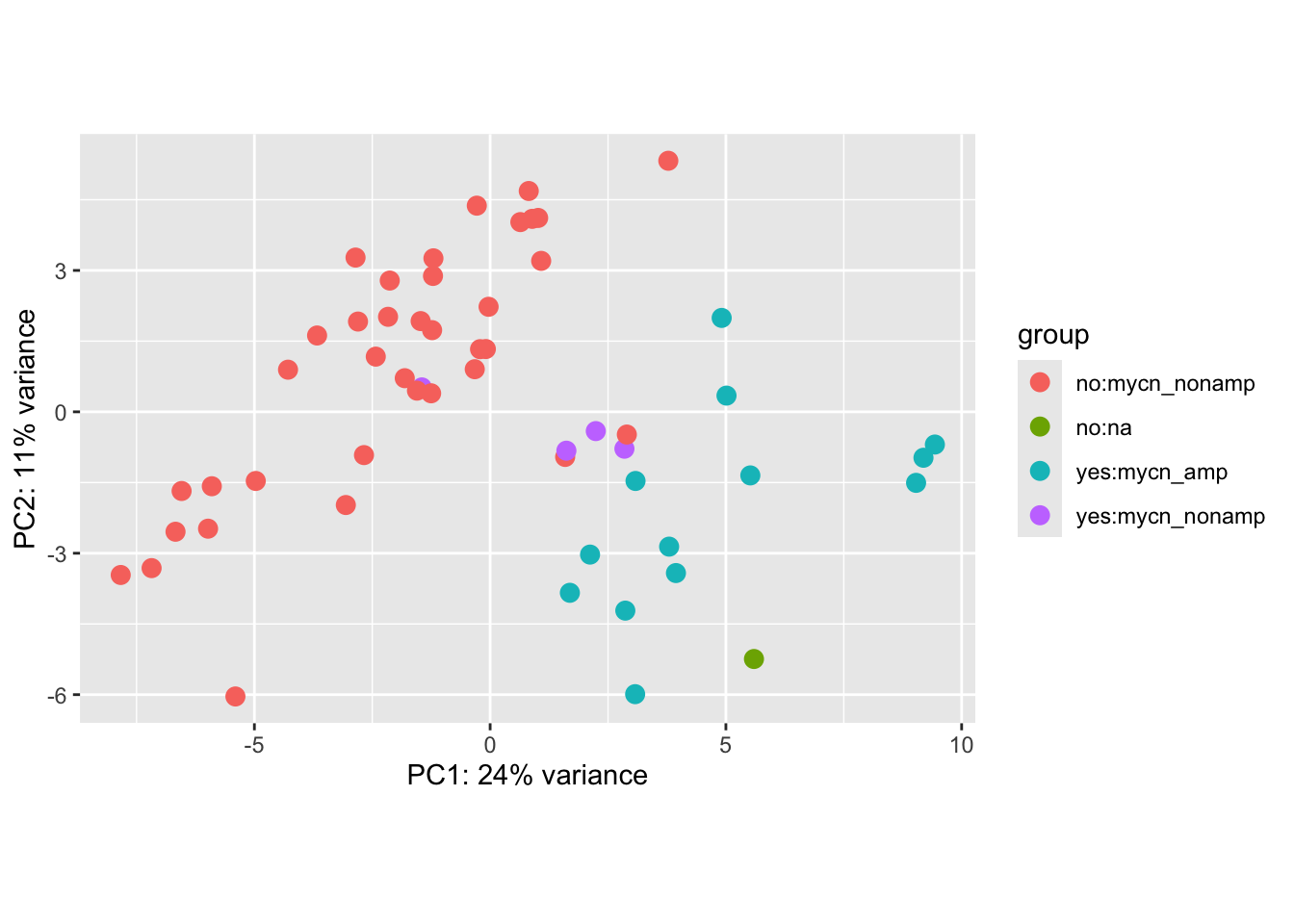
Session Information
sessionInfo()## R version 4.4.1 (2024-06-14)
## Platform: aarch64-apple-darwin20
## Running under: macOS 15.6.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/Chicago
## tzcode source: internal
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] msigdbr_7.5.1 RColorBrewer_1.1-3
## [3] pheatmap_1.0.12 clusterProfiler_4.12.6
## [5] DESeq2_1.44.0 SummarizedExperiment_1.34.0
## [7] Biobase_2.64.0 MatrixGenerics_1.16.0
## [9] matrixStats_1.4.1 GenomicRanges_1.56.1
## [11] GenomeInfoDb_1.40.1 IRanges_2.38.1
## [13] S4Vectors_0.42.1 BiocGenerics_0.50.0
## [15] readxl_1.4.3 dplyr_1.1.4
## [17] ggplot2_3.5.1 expss_0.11.6
## [19] maditr_0.8.4
##
## loaded via a namespace (and not attached):
## [1] rstudioapi_0.16.0 jsonlite_1.8.9 magrittr_2.0.3
## [4] farver_2.1.2 rmarkdown_2.28 fs_1.6.4
## [7] zlibbioc_1.50.0 vctrs_0.6.5 memoise_2.0.1
## [10] ggtree_3.12.0 htmltools_0.5.8.1 S4Arrays_1.4.1
## [13] cellranger_1.1.0 gridGraphics_0.5-1 SparseArray_1.4.8
## [16] sass_0.4.9 bslib_0.8.0 htmlwidgets_1.6.4
## [19] fontawesome_0.5.2 plyr_1.8.9 httr2_1.0.5
## [22] cachem_1.1.0 igraph_2.0.3 lifecycle_1.0.4
## [25] pkgconfig_2.0.3 gson_0.1.0 Matrix_1.7-0
## [28] R6_2.5.1 fastmap_1.2.0 GenomeInfoDbData_1.2.12
## [31] aplot_0.2.3 digest_0.6.37 enrichplot_1.24.4
## [34] colorspace_2.1-1 patchwork_1.3.0 AnnotationDbi_1.66.0
## [37] RSQLite_2.3.7 labeling_0.4.3 fansi_1.0.6
## [40] httr_1.4.7 polyclip_1.10-7 abind_1.4-8
## [43] compiler_4.4.1 bit64_4.5.2 withr_3.0.1
## [46] htmlTable_2.4.3 backports_1.5.0 BiocParallel_1.38.0
## [49] viridis_0.6.5 DBI_1.2.3 highr_0.11
## [52] ggforce_0.4.2 R.utils_2.12.3 MASS_7.3-61
## [55] rappdirs_0.3.3 DelayedArray_0.30.1 tools_4.4.1
## [58] scatterpie_0.2.4 ape_5.8 R.oo_1.26.0
## [61] glue_1.8.0 nlme_3.1-166 GOSemSim_2.30.2
## [64] shadowtext_0.1.4 grid_4.4.1 checkmate_2.3.2
## [67] reshape2_1.4.4 fgsea_1.30.0 generics_0.1.3
## [70] gtable_0.3.5 R.methodsS3_1.8.2 tidyr_1.3.1
## [73] data.table_1.16.0 tidygraph_1.3.1 utf8_1.2.4
## [76] XVector_0.44.0 ggrepel_0.9.6 pillar_1.9.0
## [79] stringr_1.5.1 babelgene_22.9 yulab.utils_0.1.7
## [82] splines_4.4.1 tweenr_2.0.3 treeio_1.28.0
## [85] lattice_0.22-6 bit_4.5.0 tidyselect_1.2.1
## [88] GO.db_3.19.1 locfit_1.5-9.10 Biostrings_2.72.1
## [91] knitr_1.48 gridExtra_2.3 xfun_0.48
## [94] graphlayouts_1.2.0 stringi_1.8.4 UCSC.utils_1.0.0
## [97] lazyeval_0.2.2 ggfun_0.1.6 yaml_2.3.10
## [100] evaluate_1.0.0 codetools_0.2-20 ggraph_2.2.1
## [103] tibble_3.2.1 qvalue_2.36.0 ggplotify_0.1.2
## [106] cli_3.6.3 munsell_0.5.1 jquerylib_0.1.4
## [109] Rcpp_1.0.13 png_0.1-8 parallel_4.4.1
## [112] blob_1.2.4 DOSE_3.30.5 tidytree_0.4.6
## [115] viridisLite_0.4.2 scales_1.3.0 purrr_1.0.2
## [118] crayon_1.5.3 rlang_1.1.4 cowplot_1.1.3
## [121] fastmatch_1.1-4 KEGGREST_1.44.1