Variant Call Pipeline
Gepoliano Chaves
September 4th, 2024
Summary
In this activity we process mitochondrial DNA FASTA sequences from self-declared white Brazilian individuals, downloaded from the GenBank. We process the FASTA sequences and then align them to the hg19 UCSC reference genome. The end results are VCF files that we need to do further processing to feed machine learning algorithms that will use the mitochondrial genetic variants to classify the individuals into their haplorgroups.
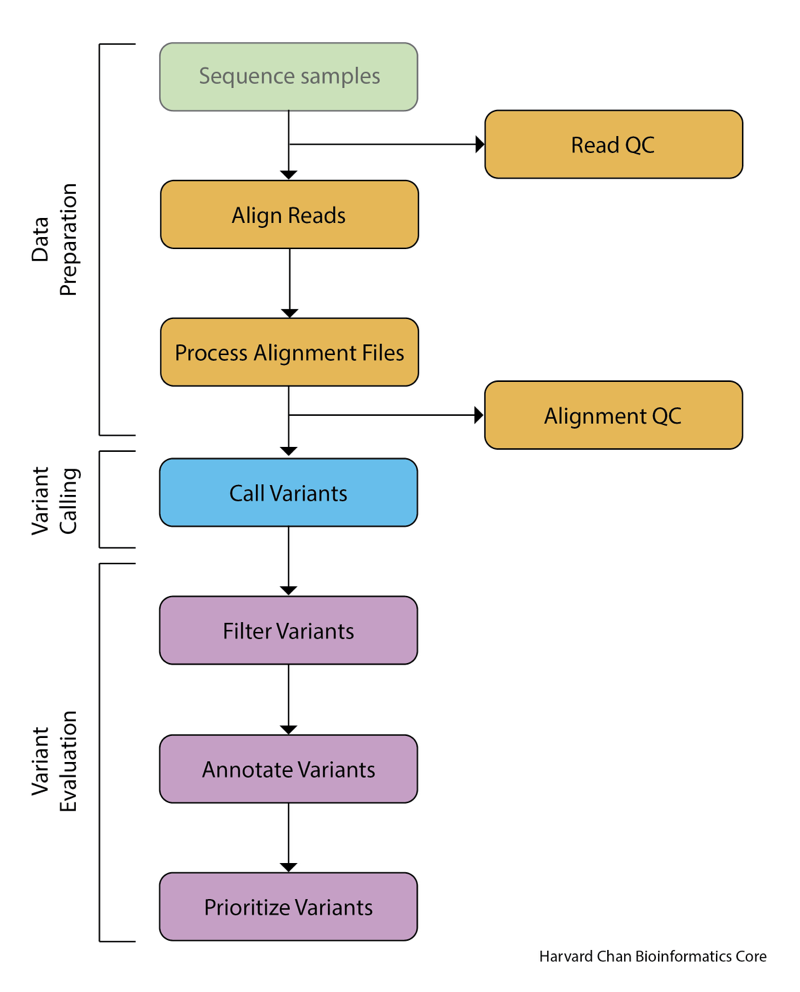
1. Process FASTA sequence
The mitochondrial sequences contained spaces in between as well as were lower case. In the first day of writing this notebook, I was not able to run those raw FASTA sequences into the first step of the variant call pipeline: alignment with BWA. This is one example of a fasta sequence that I started with:
>AF243780
tattgactca cccatcaaca accgctatgt atttcgtaca ttactgccag ccaccatgaa
tattgtacgg taccataaat acttgaccac ctgtagtaca taaaaaccca atccacatca
aaaccccctc cccatgctta caagcaagta cagcaatcaa ccctcaacta tcacacatca
actgcaactc caaagccacc cctcacccac taggatacca acaaacctac ccacccttaa
cagcacatag tacataaagc catttaccgt acatagcaca ttacagtcaa atcccttctc
gt
To fix the sequences formatting, I searched for Bash commands to remove spaces and make the letters uppercase. These were satisfying solutions I came up with:
cd ~/Desktop
string='tattgactca cccatcaaca accgctatgt atttcgtaca ttactgccag ccaccatgaa
tattgtacgg taccataaat acttgaccac ctgtagtaca taaaaaccca atccacatca
aaaccccctc cccatgctta caagcaagta cagcaatcaa ccctcaacta tcacacatca
actgcaactc caaagccacc cctcacccac taggatacca acaaacctac ccacccttaa
cagcacatag tacataaagc catttaccgt acatagcaca ttacagtcaa atcccttctc
gt'
## Remove spaces
trimmed_string=$(echo $string | tr -d ' ')
## Make uppercase
echo $trimmed_string| tr '[:lower:]' '[:upper:]' > AF243780.fasta
## Print to the first line
awk 'BEGIN { print ">AF243780" } { print }' AF243780.fasta > temp && mv temp AF243780.fastaReference for the AWK command: https://linuxconfig.org/how-to-insert-line-to-the-beginning-of-file-on-linux
Multiple fasta with capital letters and spaces removed
Finally, this part allows us to transform the mitochondrial FASTA files downloaded from the GenBank using R.
The paths are from my local machine but I transfer the capital fasta file to the appropriate folder in the hummingbird server:
After renaming the fasta sequences, they can be concatenated:
fastaDir=./data
## Change directory to the fasta folder
cd $fastaDir
## Concatenate the fasta files into one single multiple fasta sequences file.
cat AF* > mt_brazilian_exercise_multiple.fasta
cat mt_brazilian_exercise_multiple.fasta | tr -d ' ' | tr '[:lower:]' '[:upper:]' > mt_brazilian_exercise_multiple_capital.fasta
Split and rename each fasta file
Renaming the fasta sequences is necessary so the sequence names are consistent with GenBank nomenclature.
fastaDir=./data
## Change directory to the fasta folder
cd $fastaDir
## splitfasta breaks multi-fasta file
splitfasta mt_brazilian_exercise.fasta
## Change directory to the folder containing all fasta files
cd mt_brazilian_exercise_split_files
## for loop
for file in mt_brazilian_exercise*; do
echo $file;
## create variable fileName using the first line of fasta and removing the '>'
fileName=$(head -n 1 $file | sed 's/>//g')
## change the splitfasta name to the header name
mv $file $fileName".fasta"
echo "this is the new file now:" $fileName".fasta"
## move the file to the directory above.
mv $fileName".fasta" ../
done
## After everything is done, change directory to the directory above
cd ..
## Remove the filder that splitfasta created.
rm -r mt_brazilian_exercise_split_files
2. UCSC Hummingbird Server Script
Processed fasta files are transfered to the AIM with this command:
rsync -avz /hb/home/gchaves/results/geraldo/mitochondria/variant_call dcuser@ec2-3-82-97-221.compute-1.amazonaws.com:/home/dcuser
Create variant_call folder in AIM with your name
Copy the variant_call modified with your name:
cp -r variant_call variant_call_gepoliano
2.1. Index the fasta file with bwa
Example of alignment with bwa. In the references folder, run:
bwa index -a bwtsw AF243627.fasta
Example of output:
(base) dcuser@ip-172-31-90-246:~/variant_call/references$ bwa index -a bwtsw AF243627.fasta
[bwa_index] Pack FASTA... 0.00 sec
[bwa_index] Construct BWT for the packed sequence...
[BWTIncCreate] textLength=604, availableWord=65536
[bwt_gen] Finished constructing BWT in 2 iterations.
[bwa_index] 0.00 seconds elapse.
[bwa_index] Update BWT... 0.00 sec
[bwa_index] Pack forward-only FASTA... 0.00 sec
[bwa_index] Construct SA from BWT and Occ... 0.00 sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa index -a bwtsw AF243627.fasta
[main] Real time: 0.029 sec; CPU: 0.005 sec
(base) dcuser@ip-172-31-90-246:~/variant_call/references$ pwd
/home/dcuser/variant_call/references
(base) dcuser@ip-172-31-90-246:~/variant_call/references$ ls
Align reads to the index (perform for each experiment):
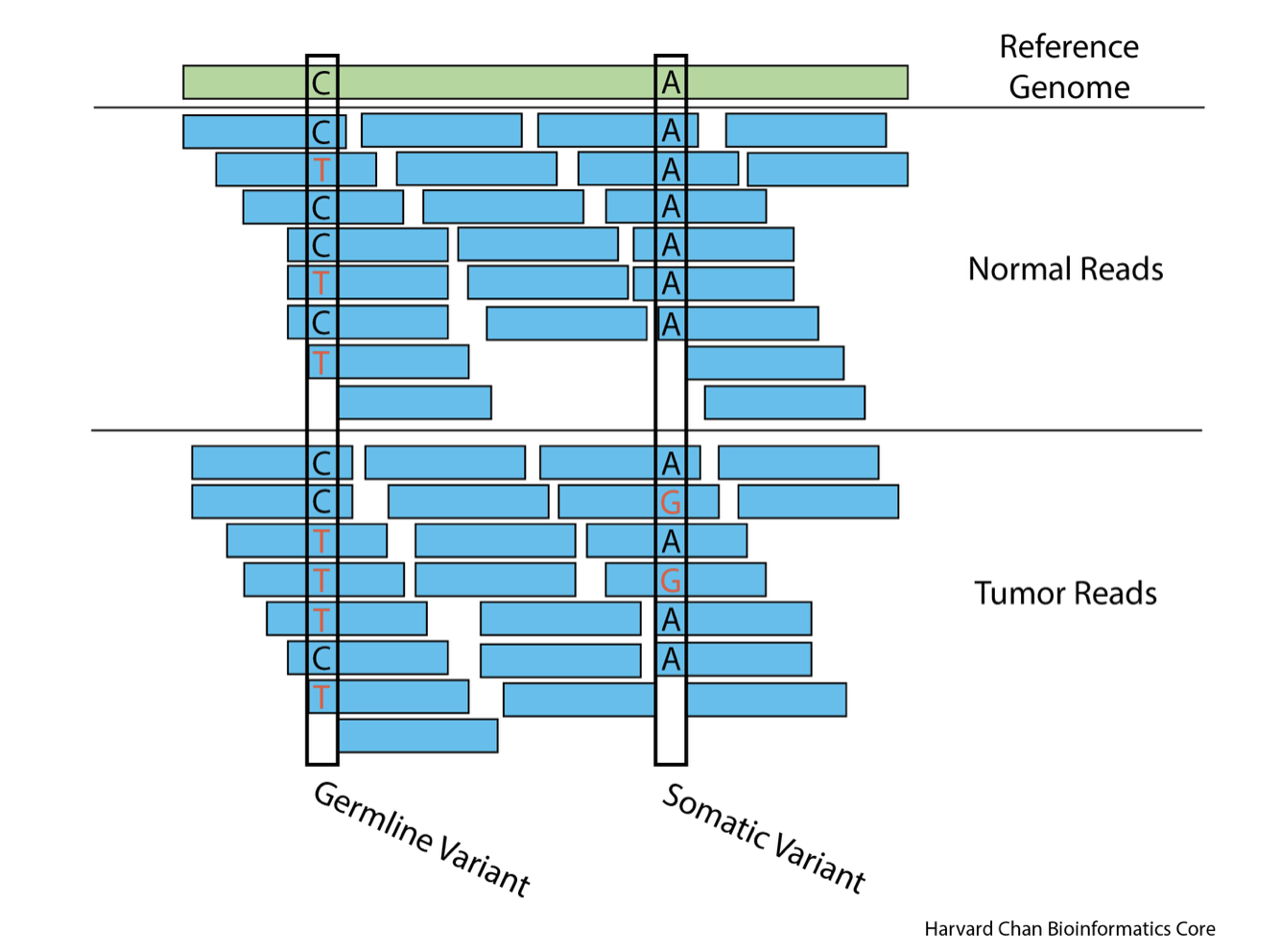
To move to the sam folder, run:
cd ../sam/
Check what exists in the sam folder:
ls ../sam/
In the sam folder, align the fasta file:
bwa mem -t 4 ../references/AF243627.fasta ../fasta/AF243628.fasta > AF243628.sam
Now check what exists in the sam folder:
ls ../sam/
There should be a result similar to this now, indicating that we have a sam (sequence alignment/map) file in the sam folder:
(base) dcuser@ip-172-31-90-246:~/variant_call/sam$ ls ../sam/
AF243628.sam
This variant call pipeline was recorded on document Lin2016/pipeline_192. In the second day of writing this notebook, I decided to run the multi-fasta file in the BWA alignment step. In the humminbird server, I store the mt_brazilian_capital.fasta in the ~/results/geraldo/mitochondria/fasta folder to run the variant call in the Scrip in Part 2: Hummingbird Script. I deleted mt_brazilian_capital.fasta in geraldo/data in my local machine because I did not want the mt_brazilian_capital.fasta to be uploaded to GitHub when publishing the geraldo website.
In this chunk I will reuse the for loop from above to run the variant call for all fasta files.
Chunk of server
This chunk is not included on purpose
Chunk of practice
In this part we assume to be inside the fasta folder.
echo "This is the task in the for loop"
REFERENCE=../references/AF243627.fasta
In this part we change directory to the fasta folder using the absolute path on purpose
We must make sure with 100% probability that we are changing the directory to the correct fasta folder
That is the reason why we must start with the absolute path to the fasta folder:
cd /home/dcuser/variant_call_gepoliano/fasta
2.2. Variant Call Pipeline using a for loop strategy
- Then we execute the Variant Call Pipeline Using a for loop Strategy:
cd /home/dcuser/variant_call_gepoliano/fasta
REFERENCE=AF243627.fasta
## for loop specific to the individual fasta files
for fastaFile in AF*.fasta; do
## Pick just the eight firt letters of the string
fastaFileName=${fastaFile:0:8}
echo "started sample $fastaFileName"
## Align
bwa mem -t 4 ../references/$REFERENCE $fastaFile > ../sam/$fastaFileName".sam"
echo "Just finished aligning sample $fastaFileName"
## SAM to BAM
samtools view -S -b ../sam/$fastaFileName".sam" > \
../bam/$fastaFileName".bam"
echo "Finished SAM to BAM of sample $fastaFileName"
## Samtools uses reference FASTA to detect "piles" in the alignment
bcftools mpileup -f ../references/$REFERENCE ../bam/$fastaFileName".bam" > \
../bcf/$fastaFileName".bcf"
## Bcftools extracts SNPs
echo "started Bcftools SNPs for sample $fastaFileName"
bcftools view -v snps ../bcf/$fastaFileName".bcf" > ../snps_vcf/$fastaFileName"_snps.vcf"
done
echo "Finished the entire for loop"
2.3. Example output of the Variant Call Pipeline:
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$ cd /home/dcuser/variant_call_gepoliano/fasta
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$ ## for loop specific to the individual fasta files
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$ for fastaFile in AF*.fasta; do
>
> ## Pick just the eight firt letters of the string
> fastaFileName=${fastaFile:0:8}
> echo "started sample $fastaFileName"
>
> ## Align
> bwa mem -t 4 $REFERENCE $fastaFile > ../sam/$fastaFileName".sam"
> echo "Just finished aligning sample $fastaFileName"
>
> ## SAM to BAM
> samtools view -S -b ../sam/$fastaFileName".sam" > \
> ../bam/$fastaFileName".bam"
> echo "Finished SAM to BAM of sample $fastaFileName"
>
> ## Samtools uses reference FASTA to detect "piles" in the alignment
> bcftools mpileup -f $REFERENCE ../bam/$fastaFileName".bam" > \
> ../bcf/$fastaFileName".bcf"
>
> ## Bcftools extracts SNPs
> echo "started Bcftools SNPs for sample $fastaFileName"
> bcftools view -v snps ../bcf/$fastaFileName".bcf" > ../snps_vcf/$fastaFileName"_snps.vcf"
>
> done
started sample AF243628
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243628.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243628
Finished SAM to BAM of sample AF243628
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243628
started sample AF243629
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243629.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243629
Finished SAM to BAM of sample AF243629
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243629
started sample AF243630
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243630.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243630
Finished SAM to BAM of sample AF243630
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243630
started sample AF243631
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243631.fasta
[main] Real time: 0.007 sec; CPU: 0.003 sec
Just finished aligning sample AF243631
Finished SAM to BAM of sample AF243631
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243631
started sample AF243632
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243632.fasta
[main] Real time: 0.004 sec; CPU: 0.002 sec
Just finished aligning sample AF243632
Finished SAM to BAM of sample AF243632
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243632
started sample AF243633
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.002 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243633.fasta
[main] Real time: 0.008 sec; CPU: 0.004 sec
Just finished aligning sample AF243633
Finished SAM to BAM of sample AF243633
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243633
started sample AF243700
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243700.fasta
[main] Real time: 0.007 sec; CPU: 0.003 sec
Just finished aligning sample AF243700
Finished SAM to BAM of sample AF243700
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243700
started sample AF243701
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243701.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243701
Finished SAM to BAM of sample AF243701
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243701
started sample AF243702
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243702.fasta
[main] Real time: 0.006 sec; CPU: 0.003 sec
Just finished aligning sample AF243702
Finished SAM to BAM of sample AF243702
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243702
started sample AF243703
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243703.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243703
Finished SAM to BAM of sample AF243703
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243703
started sample AF243704
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.002 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243704.fasta
[main] Real time: 0.006 sec; CPU: 0.004 sec
Just finished aligning sample AF243704
Finished SAM to BAM of sample AF243704
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243704
started sample AF243705
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243705.fasta
[main] Real time: 0.006 sec; CPU: 0.003 sec
Just finished aligning sample AF243705
Finished SAM to BAM of sample AF243705
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243705
started sample AF243706
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243706.fasta
[main] Real time: 0.004 sec; CPU: 0.002 sec
Just finished aligning sample AF243706
Finished SAM to BAM of sample AF243706
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243706
started sample AF243707
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243707.fasta
[main] Real time: 0.005 sec; CPU: 0.004 sec
Just finished aligning sample AF243707
Finished SAM to BAM of sample AF243707
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243707
started sample AF243708
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243708.fasta
[main] Real time: 0.006 sec; CPU: 0.003 sec
Just finished aligning sample AF243708
Finished SAM to BAM of sample AF243708
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243708
started sample AF243709
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (301 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.002 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243709.fasta
[main] Real time: 0.007 sec; CPU: 0.003 sec
Just finished aligning sample AF243709
Finished SAM to BAM of sample AF243709
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243709
started sample AF243780
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243780.fasta
[main] Real time: 0.006 sec; CPU: 0.003 sec
Just finished aligning sample AF243780
Finished SAM to BAM of sample AF243780
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243780
started sample AF243781
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243781.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243781
Finished SAM to BAM of sample AF243781
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243781
started sample AF243782
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243782.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243782
Finished SAM to BAM of sample AF243782
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243782
started sample AF243783
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243783.fasta
[main] Real time: 0.006 sec; CPU: 0.002 sec
Just finished aligning sample AF243783
Finished SAM to BAM of sample AF243783
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243783
started sample AF243784
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243784.fasta
[main] Real time: 0.003 sec; CPU: 0.002 sec
Just finished aligning sample AF243784
Finished SAM to BAM of sample AF243784
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243784
started sample AF243785
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.000 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243785.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243785
Finished SAM to BAM of sample AF243785
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243785
started sample AF243786
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243786.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243786
Finished SAM to BAM of sample AF243786
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243786
started sample AF243787
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243787.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243787
Finished SAM to BAM of sample AF243787
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243787
started sample AF243788
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (301 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243788.fasta
[main] Real time: 0.003 sec; CPU: 0.002 sec
Just finished aligning sample AF243788
Finished SAM to BAM of sample AF243788
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243788
started sample AF243789
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.000 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243789.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243789
Finished SAM to BAM of sample AF243789
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243789
started sample AF243790
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243790.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243790
Finished SAM to BAM of sample AF243790
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243790
started sample AF243791
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243791.fasta
[main] Real time: 0.004 sec; CPU: 0.003 sec
Just finished aligning sample AF243791
Finished SAM to BAM of sample AF243791
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243791
started sample AF243792
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243792.fasta
[main] Real time: 0.005 sec; CPU: 0.003 sec
Just finished aligning sample AF243792
Finished SAM to BAM of sample AF243792
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243792
started sample AF243793
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243793.fasta
[main] Real time: 0.004 sec; CPU: 0.003 sec
Just finished aligning sample AF243793
Finished SAM to BAM of sample AF243793
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243793
started sample AF243794
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.002 CPU sec, 0.002 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243794.fasta
[main] Real time: 0.007 sec; CPU: 0.004 sec
Just finished aligning sample AF243794
Finished SAM to BAM of sample AF243794
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243794
started sample AF243795
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243795.fasta
[main] Real time: 0.005 sec; CPU: 0.002 sec
Just finished aligning sample AF243795
Finished SAM to BAM of sample AF243795
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243795
started sample AF243796
[M::bwa_idx_load_from_disk] read 0 ALT contigs
[M::process] read 1 sequences (302 bp)...
[M::mem_process_seqs] Processed 1 reads in 0.001 CPU sec, 0.001 real sec
[main] Version: 0.7.17-r1188
[main] CMD: bwa mem -t 4 ../references/AF243627.fasta AF243796.fasta
[main] Real time: 0.006 sec; CPU: 0.003 sec
Just finished aligning sample AF243796
Finished SAM to BAM of sample AF243796
[mpileup] 1 samples in 1 input files
started Bcftools SNPs for sample AF243796
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$
(base) dcuser@ip-172-31-90-246:~/variant_call_gepoliano/fasta$ echo "Finished the entire for loop"
Finished the entire for loop
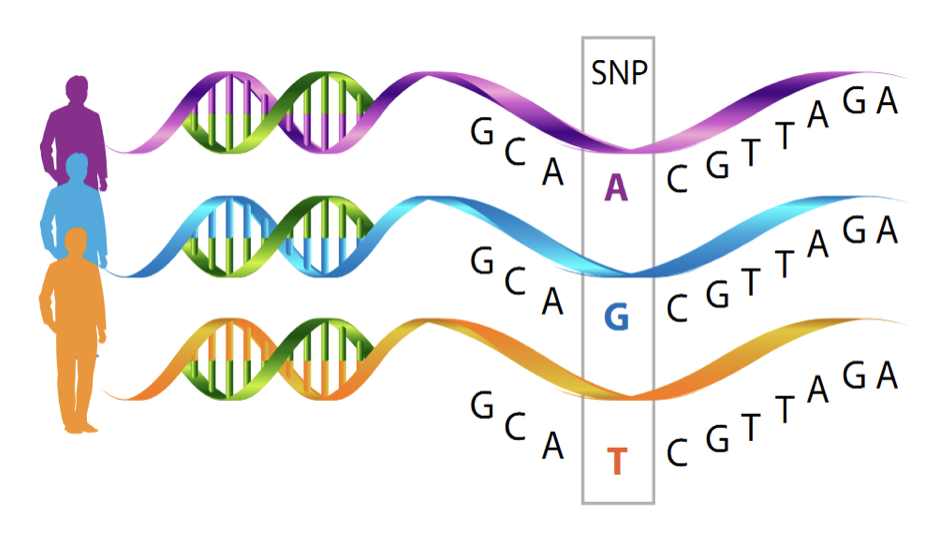
Fix the bcftools error
After a couple hours dealing with this error:
Failed to read from /hb/home/gchaves/results/geraldo/mitochondria/bcf/AF243782.bcf: unknown file type
I noticed this part in the error message:
Note that using "samtools mpileup" to generate BCF or VCF files has been
removed. To output these formats, please use "bcftools mpileup" instead.
I then replaced the line in the code above, to
bcftools mpileup -f ~/references/hg19/GRCh37.p13.genome.fa ~/results/geraldo/mitochondria/bam/AF243780.bam > ~/results/geraldo/mitochondria/bcf/AF243780.bcf
References
https://hbctraining.github.io/variant_analysis/lessons/00_intro_to_variant_calling.html https://stackoverflow.com/questions/2439579/how-to-get-the-first-line-of-a-file-in-a-bash-script https://unix.stackexchange.com/questions/104881/remove-particular-characters-from-a-variable-using-bash https://linuxsimply.com/bash-scripting-tutorial/string/substring/